vaspparser demo#
This notebook walks through the main building blocks of the vaspparser package using the
sample VASP output files that ship with the test suite (tests/static/vasp_test_files).
It covers:
Reading and writing VASP structure files (
POSCAR/CONTCAR)Parsing a full VASP run with
parse_vasp_outputParsing individual files:
OSZICAR,OUTCAR,vasprun.xml,PROCARWorking with the electronic structure: density of states (DOS) and the band gap
Reading volumetric data (
CHGCAR)Bader charge analysis helpers
import os
import numpy as np
# Sample VASP output files used throughout the test suite
TEST_FILES = os.path.join("static", "vasp_test_files")
FULL_JOB_SAMPLE = os.path.join(TEST_FILES, "full_job_sample")
1. Reading and writing structures#
VASP structures are stored in the POSCAR/CONTCAR format. read_atoms parses such a file into an ASE Atoms object, and write_poscar does the reverse.
from vaspparser.vasp.structure import read_atoms, write_poscar, vasp_sorter
structure = read_atoms(os.path.join(FULL_JOB_SAMPLE, "POSCAR"), species_list=["Fe"])
structure
Atoms(symbols='Fe2', pbc=True, cell=[2.8, 2.8, 2.8])
# vasp_sorter() gives the permutation that groups atoms by species, which is how VASP
# expects atoms to be ordered inside a POSCAR file
vasp_sorter(structure)
array([0, 1])
write_poscar(structure=structure, filename="POSCAR_demo")
roundtrip = read_atoms("POSCAR_demo")
assert roundtrip == structure
os.remove("POSCAR_demo")
roundtrip
Atoms(symbols='Fe2', pbc=True, cell=[2.8, 2.8, 2.8])
2. Parsing a full VASP run#
parse_vasp_output is the main entry point of the package: point it at a calculation
directory and it returns a single, hierarchical dictionary with everything that could be
parsed (energies, forces, structure, electronic structure, charge density, …).
from vaspparser.vasp.output import parse_vasp_output
output_dict = parse_vasp_output(working_directory=FULL_JOB_SAMPLE)
list(output_dict.keys())
['description',
'generic',
'structure',
'charge_density',
'electronic_structure',
'outcar']
# Generic, code-agnostic quantities (energies, forces, positions, ...)
list(output_dict["generic"].keys())
['temperature',
'stresses',
'pressures',
'elastic_constants',
'forces',
'cells',
'volume',
'energy_pot',
'energy_tot',
'steps',
'positions',
'dft']
energy_tot = output_dict["generic"]["energy_tot"]
print(f"Number of ionic steps: {len(energy_tot)}")
print(f"Final total energy: {energy_tot[-1]:.6f} eV")
Number of ionic steps: 1
Final total energy: -17.737987 eV
3. Parsing individual output files#
Each VASP file also has a dedicated parser that can be used on its own.
OSZICAR#
The OSZICAR file tracks the energy at every electronic/ionic step.
from vaspparser.vasp.parser.oszicar import Oszicar
oszicar = Oszicar()
oszicar.from_file(filename=os.path.join(FULL_JOB_SAMPLE, "OSZICAR"))
oszicar.parse_dict["energy_pot"]
array([-17.73798679])
OUTCAR#
The OUTCAR file contains most of what VASP printed during the run: energies, forces, stresses, the Fermi level, magnetic moments, and more.
from vaspparser.vasp.parser.outcar import Outcar
outcar = Outcar()
outcar.from_file(filename=os.path.join(FULL_JOB_SAMPLE, "OUTCAR"))
print("Fermi level (eV):", outcar.parse_dict["fermi_level"])
print("Number of electrons:", outcar.parse_dict["n_elect"])
print("Final pressure:", outcar.parse_dict["pressures"][-1])
Fermi level (eV): 5.9788
Number of electrons: 16.0
Final pressure: [-0.08177043 -0.08177043 -0.08177043]
vasprun.xml#
vasprun.xml is VASP’s structured XML output and is the preferred source for energies, forces, and the electronic structure when available.
from vaspparser.vasp.vasprun import Vasprun
vasprun_path = os.path.join(TEST_FILES, "vasprun_samples", "vasprun_2.xml")
vasprun = Vasprun()
vasprun.from_file(filename=vasprun_path)
print("Final energy (eV):", vasprun.vasprun_dict["total_fr_energies"][-1])
Final energy (eV): -67.64570454
4. Electronic structure: DOS and band gap#
Vasprun.get_electronic_structure() returns an ElectronicStructure object with the
eigenvalues/occupancies for every k-point and spin channel. From there we can compute the
density of states and the band gap.
electronic_structure = vasprun.get_electronic_structure()
print("Eigenvalue matrix shape (spin, kpoints, bands):", electronic_structure.eigenvalue_matrix.shape)
electronic_structure.get_band_gap()
Eigenvalue matrix shape (spin, kpoints, bands): (1, 36, 72)
{0: {'band_gap': np.float64(0.007800000000000001),
'vbm': {'value': np.float64(-0.1611),
'kpoint': <vaspparser.dft.waves.electronic.Kpoint at 0x7544a5a8b4d0>,
'band': <vaspparser.dft.waves.electronic.Band at 0x7544a594fb30>},
'cbm': {'value': np.float64(-0.1533),
'kpoint': <vaspparser.dft.waves.electronic.Kpoint at 0x7544a5a8b1c0>,
'band': <vaspparser.dft.waves.electronic.Band at 0x7544a5908230>}}}
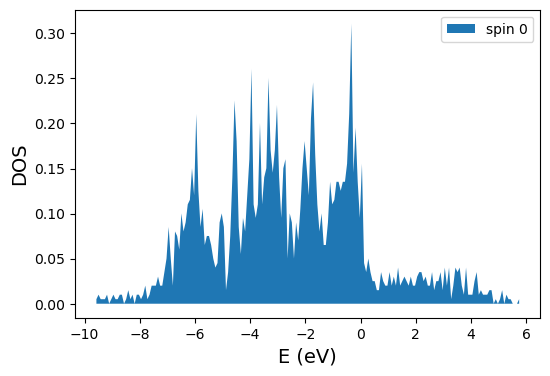
from vaspparser.dft.waves.dos import Dos
dos = Dos(es_obj=electronic_structure, n_bins=200)
plot = dos.plot_total_dos()
plot.show()
PROCAR#
The PROCAR file holds atom- and orbital-projected band/DOS information, useful for analysing the chemical character of bands.
from vaspparser.vasp.procar import Procar
procar = Procar()
procar_es = procar.from_file(filename=os.path.join(TEST_FILES, "PROCAR_for_test"))
band = procar_es.kpoints[0].bands[0][0]
print("Eigenvalue:", band.eigenvalue, "Occupancy:", band.occupancy)
print("Per-atom resolved DOS:", band.atom_resolved_dos)
Eigenvalue: -17.37867948 Occupancy: 1.0
Per-atom resolved DOS: [0.145 0.298 0.298]
5. Volumetric data: charge density#
CHGCAR/LOCPOT-style files describe a quantity on a real-space grid. VaspVolumetricData
parses them and offers helpers such as averaging the density along one lattice direction
(handy for e.g. work-function or interface analysis).
from vaspparser.vasp.volumetric_data import VaspVolumetricData
charge_density = VaspVolumetricData()
charge_density.from_file(filename=os.path.join(FULL_JOB_SAMPLE, "CHGCAR"), normalize=True)
print("Grid shape:", charge_density.total_data.shape)
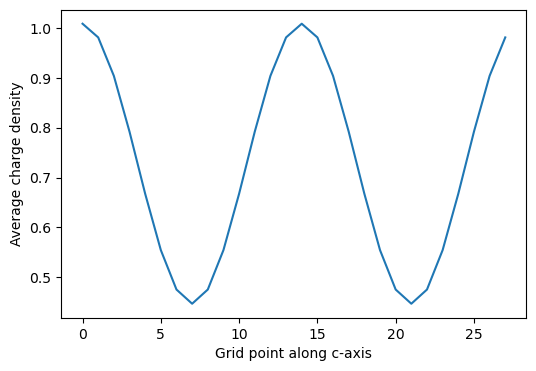
average_along_c = charge_density.get_average_along_axis(ind=2)
average_along_c
Grid shape: (28, 28, 28)
array([1.00830804, 0.98088211, 0.90377918, 0.79195173, 0.66754708,
0.55462055, 0.47555225, 0.44711719, 0.47555225, 0.55462055,
0.66754708, 0.79195173, 0.90377918, 0.98088211, 1.00830804,
0.98088211, 0.90377918, 0.79195173, 0.66754708, 0.55462055,
0.47555225, 0.44711719, 0.47555225, 0.55462055, 0.66754708,
0.79195173, 0.90377918, 0.98088211])
import matplotlib.pyplot as plt
plt.figure(figsize=(6, 4))
plt.plot(average_along_c)
plt.xlabel("Grid point along c-axis")
plt.ylabel("Average charge density")
plt.show()
6. Bader charge analysis#
Bader analysis partitions the charge density into atomic basins. Running the actual
analysis requires the external bader executable, but vaspparser also exposes helpers
to parse its output (ACF.dat) and to combine AECCAR0/AECCAR2 into the valence and
total charge density used as the Bader input.
from vaspparser.dft.bader import parse_charge_vol_file, get_valence_and_total_charge_density
bader_test_path = os.path.join(TEST_FILES, "bader_test")
bader_structure = read_atoms(os.path.join(bader_test_path, "POSCAR"))
acf_file = os.path.join("static", "dft", "bader_files", "ACF.dat")
charges, volumes = parse_charge_vol_file(structure=bader_structure, filename=acf_file)
print("Bader charges:", charges)
print("Bader volumes:", volumes)
Bader charges: [0.438202 0.438197 7.143794]
Bader volumes: [287.28469 297.577878 415.155432]
charge_density_valence, charge_density_total = get_valence_and_total_charge_density(
working_directory=bader_test_path
)
print("Valence charge density grid:", charge_density_valence.total_data.shape)
print("Total (core + valence) charge density grid:", charge_density_total.total_data.shape)
Valence charge density grid: (20, 20, 20)
Total (core + valence) charge density grid: (20, 20, 20)